 EpHod
EpHod
# Machine learning prediction of enzyme optimum pH
Journal: Nature Machine Intelligence (IF 23.9) Published: 2025年4月29日 开源地址:https://github.com/beckham-lab/EpHod
# 模型结构
EpHod并不是一个单一的模型,而是一个集成模型(Ensemble Model)。它结合了两种利用相同底层特征(ESM-1v嵌入)但架构不同的模型,通过平均它们的预测结果来提升准确性和稳健性。
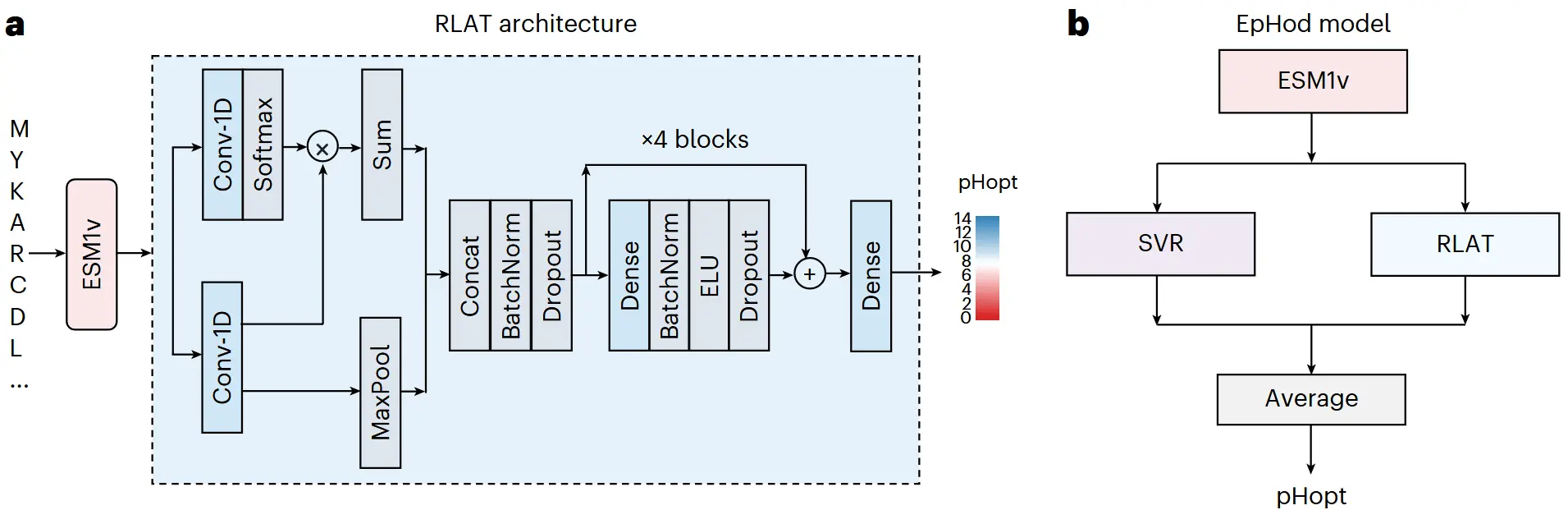
EpHod的两个核心组件是:
1. 支持向量回归模型 (ESM-1v-SVR)
- 模型类型:传统的机器学习模型(Support Vector Regression)。SVR擅长处理高维数据,并能通过核函数技巧学习非线性关系。
- 输入特征:**平均化(Averaged)**的ESM-1v嵌入。具体做法是,将一个蛋白质序列中每个氨基酸的ESM-1v嵌入向量(例如1280维)进行逐维度平均,最终得到一个代表整个蛋白质的单一向量。
- 作用:这个分支捕捉了蛋白质序列全局的、整体性的生物化学信息。它的结构相对简单,训练速度快,且在全局特征表示上表现非常出色。
2. 残差光注意力网络(ESM-1v-RLATtr)
- 模型类型:深度学习神经网络(Residual Light Attention Network)。
- 输入特征:**逐残基(Per-residue)**的ESM-1v嵌入。它直接使用蛋白质序列每个位置上氨基酸的嵌入向量,形成一个二维的张量。
- **模型架构 **:
- Light Attention:与传统的Transformer注意力机制不同,它使用两个并行的1D卷积层来分别学习每个位置的“重要性”(注意力权重)和“值”(转换后的特征),然后加权求和。这是一种更轻量、高效的注意力机制。
- Residual Blocks:模型的核心由多个残差块堆叠而成。这意味着网络的层数可以很深,从而学习更复杂的特征映射,同时残差连接可以有效防止梯度消失问题,使深度网络更容易训练。
- 作用:这个分支能够捕捉局部和位置相关的信息。通过注意力机制,它能自主学习到序列中哪些氨基酸(例如催化位点附近、蛋白质表面的带电残基)对最终的pHopt预测贡献最大。这是模型可解释性的关键来源。
集成方式:将SVR模型和RLATtr模型对同一个蛋白质序列的pHopt预测值进行简单的算术平均,得到最终的EpHod预测结果。集成学习通常能降低单一模型的偏差和方差,从而获得更稳定和准确的预测。
# 损失函数
该研究使用的核心损失函数是加权均-方根误差 (reweighted RMSE) 。
# 1. 基础:均方根误差 (RMSE)
我们先从标准RMSE的公式开始,它衡量的是模型预测值与真实值之间的差异。
- 是第 个样本的真实pH值。
- 是模型对第 个样本预测的pH值。
- 是样本总数。
这个函数计算误差的平方的平均值,然后开方。开方的目的是让误差的单位与原始数据的单位(即pH单位)保持一致,从而更直观地理解误差的大小。
# 2. 动机:为什么要“加权”?
论文中反复强调,他们使用的数据集存在严重的标签分布不均衡问题。
- 问题所在:数据集中将近75%的酶的最适pH值 () 集中在6到8之间,而极端酸性 (pH < 5) 和极端碱性 (pH > 9) 的酶样本非常稀少。
# 3. 解决方案:加权损失函数
为了解决这个问题,研究者为每个样本引入了一个权重 ()。这个权重的核心思想是:样本越稀有,权重越高;样本越常见,权重越低。 论文中使用的加权RMSE公式如下:
# 公式详解:
- 和 :同上,分别是真实值和预测值。
- :指每个训练批次 (batch) 的大小,而不是整个数据集的大小。
- :代表经过均值归一化的样本权重 。
# 权重的计算方法 ( ):
计算基础权重 (): 论文评估了五种不同的方法来计算每个样本的基础权重,其中最直观的一种叫做 "bin inverse"(分箱倒数)。
- 步骤1:分箱。将所有pH值分为三个区间(箱):酸性 (
pH ≤ 5)、中性 (5 < pH < 9) 和碱性 (pH ≥ 9) 。 - 步骤2:计算倒数。对于某个箱内的任何一个样本,其权重 就是该箱内样本总数的倒数。例如,如果碱性箱里只有100个样本,而中性箱里有7000个样本,那么一个碱性样本的权重就是 ,而一个中性样本的权重是 。显然,碱性样本的权重远大于中性样本。
- 步骤1:分箱。将所有pH值分为三个区间(箱):酸性 (
归一化权重 (): 直接使用基础权重可能会导致梯度(模型更新的依据)的方差过大,使训练不稳定。因此,论文对权重进行了均值归一化处理 。
这个操作将所有权重的平均值调整为1,同时保持了它们之间的相对比例,有助于稳定训练过程 。
# 4. 工作机制总结
通过这个加权损失函数,训练过程发生了如下变化:
- 当模型预测一个中性pH的酶时,即使预测出现了一点误差,由于其权重 很小,对总损失 (loss) 的贡献也较小。
- 当模型预测一个稀有的碱性pH的酶时,如果预测出现同样的误差,由于其权重 很大,这个误差会被放大,对总损失的贡献也会变得很大。
- 为了最小化总损失,优化算法(如Adam)会迫使模型参数进行更大的调整,来修正对稀有样本的预测错误。
最终,这种方法有效地引导模型去关注那些数量稀少但十分重要的极端pH值样本,显著提升了模型在酸性和碱性酶上的预测准确性,使其更具生物技术应用价值。