 LigandMPNN
LigandMPNN
# LigandMPNN ( Nature Methods 2025 IF 36.1 )
 传统蛋白质序列设计方法(如 Rosetta 和 ProteinMPNN)主要专注于蛋白质自身的结构,而 忽略了小分子、核酸和金属离子等非蛋白原子上下文,这对于酶、传感器和小分子结合蛋白的设计至关重要。
LigandMPNN 首次显式引入非蛋白原子上下文(如小分子、核酸、金属),用于蛋白质序列与侧链构象设计。
LigandMPNN 已被用于设计 超过100种经过实验验证的小分子和DNA结合蛋白,这些蛋白具有高亲和力和高结构准确性(其中四种通过X射线晶体结构得到验证)。
传统蛋白质序列设计方法(如 Rosetta 和 ProteinMPNN)主要专注于蛋白质自身的结构,而 忽略了小分子、核酸和金属离子等非蛋白原子上下文,这对于酶、传感器和小分子结合蛋白的设计至关重要。
LigandMPNN 首次显式引入非蛋白原子上下文(如小分子、核酸、金属),用于蛋白质序列与侧链构象设计。
LigandMPNN 已被用于设计 超过100种经过实验验证的小分子和DNA结合蛋白,这些蛋白具有高亲和力和高结构准确性(其中四种通过X射线晶体结构得到验证)。
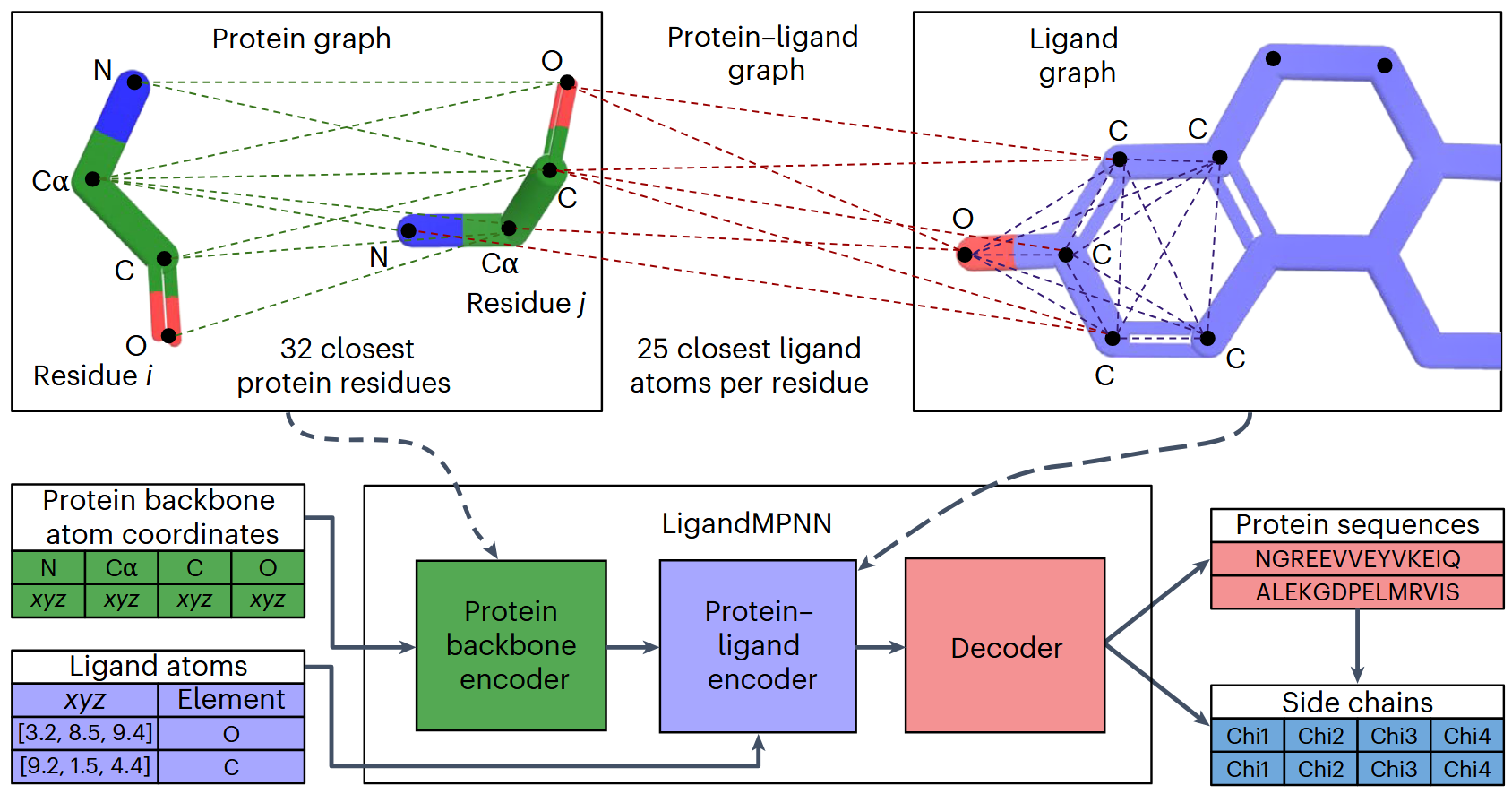
# 输入
LigandMPNN 的输入由 蛋白主链信息 + 非蛋白上下文信息(即配体原子) 两部分组成,形式化定义如下:
# 1. 蛋白主链坐标 X
- 表示为
- 每个残基提供 4 个原子坐标:N、Cα、C、O
- 总共有 个残基(长度为 L 的蛋白)
# 2. 非蛋白上下文坐标 Y(配体原子)
- 表示为
- 对每个残基选取其附近最近的 个配体原子(如小分子或金属原子)
# 3. 配体原子掩码 Y_m
- 表示为
- 掩码标记哪些配体原子是真实存在的,哪些是填充的
[!note] 在真实蛋白–配体结构中,不同残基周围的配体原子数量可能不同,例如: 有的残基周围可能只有 12 个原子(距离较远或稀疏); 有的残基周围可能有 30 个原子(配体大,或靠近结合位点)。 为了实现并行计算,需要固定大小的输入张量。所以:
# 4. 配体原子类型 Y_t
- 表示为
- 每个配体原子是一个化学元素(如 C、N、O、Zn 等),通常是 one-hot 编码
# 上述输入如何特征化的

# Algorithm 10:Protein and ligand featurization
该算法的作用是:
- 为 蛋白质残基图 构造边特征;(proteinMPNN一样的)
- 为 配体原子图 和 蛋白–配体图 构造节点与边特征;(独有的)
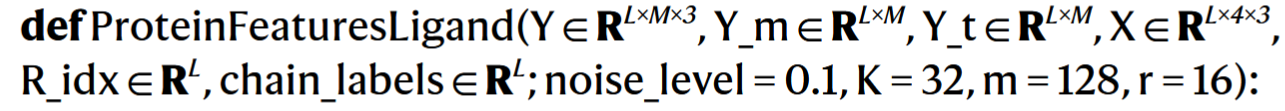
| 名称 | 描述 |
|---|---|
Y ∈ ℝ^{L×M×3} | 每个残基最近的 M 个配体原子坐标 |
Y_m ∈ ℝ^{L×M} | 配体掩码 |
Y_t ∈ ℝ^{L×M} | 配体原子类型编号 |
X ∈ ℝ^{L×4×3} | 蛋白质主链 4 个原子的坐标(N, Cα, C, O) |
R_idx ∈ ℝ^L | 残基编号 |
chain_labels ∈ ℝ^L | 所属链编号 |
noise_level | 控制噪声强度 |
K | 每个残基邻接的蛋白质节点数(默认 32) |
m | 最终输出特征维度 |
r | RBF 编码维度,默认16,和proteinMPNN一样 |
# 1. 坐标扰动(抗过拟合)
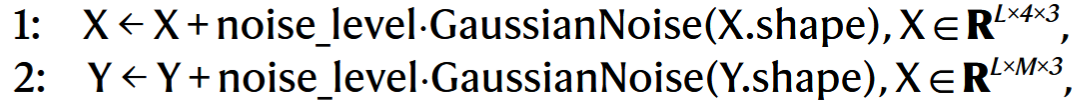
# 2. 构建虚拟 Cβ 原子(共 5 个 backbone 原子)
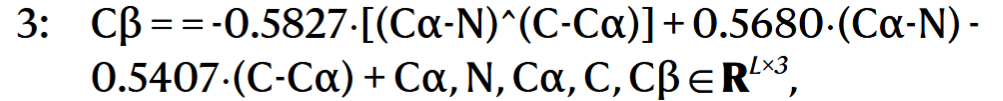
# 3. 构造蛋白质图的邻接关系(top-K)
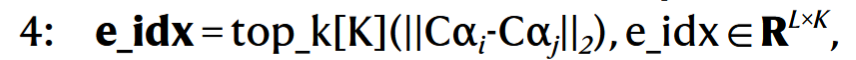
- 基于 Cα–Cα 距离构建最近邻图(残基–残基);
- 每个残基连接 K 个邻居。
# 4. 构造蛋白质图的距离边特征(RBF 编码)

- 所有 5×5 = 25 对 backbone 原子对之间距离;
- 每个距离用 r 维 RBF 编码;
- 最终特征维度为 25×r。
# 5. 序列位置编码特征(位置差 + 是否同链)

- 通过位置差和是否同链得到位置编码
# 6. 根据距离特征和位置编码特征得到最终的边特征
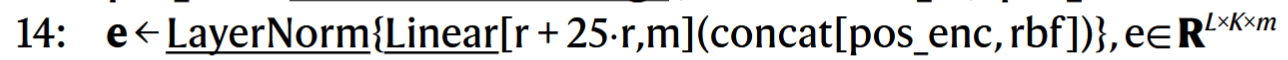
- 附加一个降维操作,
25⋅r + r → m
# 7. 配体原子类型嵌入(增强化学表示)
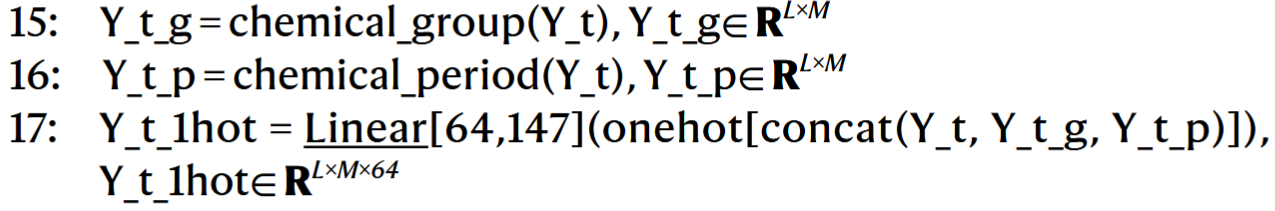
Y_t 配体原子类型。编码元素周期表中元素的族、周期;
# 8. 构造蛋白–配体图的距离边特征
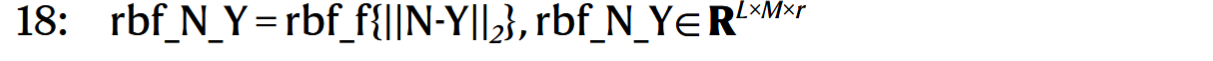
 计算每个 backbone 原子(N, Cα, C, O, Cβ)到其对应 某个配体原子的距离,然后使用RBF编码,然后concat起来。
计算每个 backbone 原子(N, Cα, C, O, Cβ)到其对应 某个配体原子的距离,然后使用RBF编码,然后concat起来。
# 9. 构造蛋白–配体图的角度特征(N–Cα–C 构建坐标系)
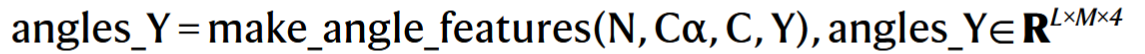 根据N–Cα–C和配体原子Y构建角度特征。
根据N–Cα–C和配体原子Y构建角度特征。
# 10. 根据距离特征、配体原子、角度特征构造配体节点初始特征
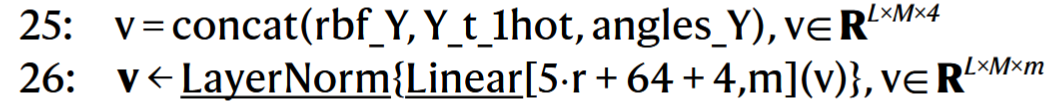 将一个配体与一个氨基酸的RBF特征、自身的化学特征编码和角度特征一起得到该配体节点的特征
将一个配体与一个氨基酸的RBF特征、自身的化学特征编码和角度特征一起得到该配体节点的特征
# 11. 构造配体图的边特征
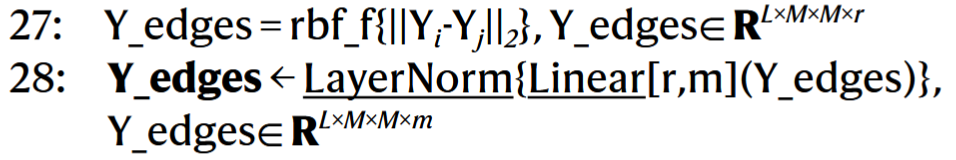 通过配体与配体之间的距离来得到边特征
通过配体与配体之间的距离来得到边特征
# 12. 构造配体图的节点特征
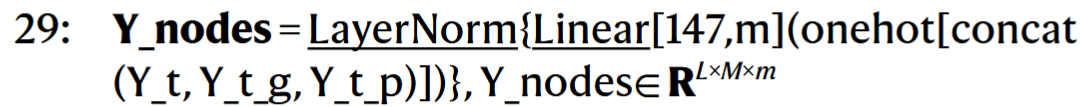
Y_t是原子类型,Y_t_g和Y_t_p是原子的族和周期信息。
# 13. 输出
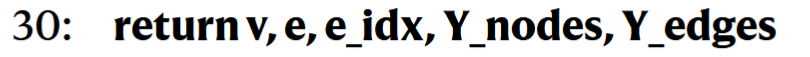
| 名称 | 维度 | 说明 |
|---|---|---|
v | [L×M×m] | 蛋白–配体图中配体节点表示 |
e | [L×K×m] | 蛋白质残基图中的边特征 |
e_idx | [L×K] | 残基邻接索引,top-K |
Y_nodes | [L×M×m] | 配体图中原子节点特征 |
Y_edges | [L×M×M×m] | 配体图中边特征(全连接) |
# 输出(Output)
LigandMPNN 的输出包含两个核心部分:
# 1. 氨基酸序列
- 形式:长度为 LLL 的氨基酸序列
- 通过 自回归方式逐个位点生成
- 每个位点输出一个 20 类别 softmax(对应 20 种标准氨基酸)
- 支持温度采样(temperature sampling)以调节多样性
# 2. 侧链构象
- 对每个残基,预测侧链的构象角:chi1, chi2, chi3, chi4。并没有直接预测这四个角。
- 输出为 每个角度的三混合分布(Mixture of Circular Normal):
- 每个 chi 角度预测 3 个高斯成分(均值、方差、混合权重)
[!note] 背景知识:侧链 χ 角 & Von Mises 分布
- 每个氨基酸残基的侧链由 1 到 4 个扭转角(χ₁~χ₄)决定;
- 它们是 圆形变量,值域是 ;
- 用正态分布建模会忽略角度的周期性,而 Von Mises 分布(环状高斯)非常适合建模这种类型;
# 模型架构
# 1. Encoder Layer
和proteinMPNN一样的,用于将蛋白质残基图中的边特征e和一个初始为零的节点特征v,更新出来新的边特征和节点特征。
v表示残基节点,e表示残基与残基之间的边。

- 边消息构造(节点 + 边特征拼接)
- 边消息聚合(邻接边求和)
- 更新节点
- 类似的方式更新边
# 2. Decoder Layer
和proteinMPNN类似的,给出节点特征和边特征来更新节点特征。节点特征来得到最后的预测。
v表示残基节点,e表示残基与残基之间的边。

- 边消息构造(节点 + 边特征拼接)
- 边消息聚合(邻接边求和)
- 更新节点
# 3. Context Decoder Layer
这个是LigandMPNN新提出来的,主要用于在“配体原子图”中进行节点信息更新。不过和Decoder Layer很类似。
v表示配体节点,e表示配体与配体之间的边。

- 对于每个残基 下的配体原子 和它的邻居原子 ,将:
- 当前节点特征
- 边特征 拼接成一条边消息。
- 然后聚合更新配体边节点
# 4. LigandMPNN encode function
有了每个 layer 的定义就可以看看具体的 encoder 里面了。
作用就是融合三种图,得到融合后的蛋白质图的边特征和节点特征。 
# 0. 函数定义
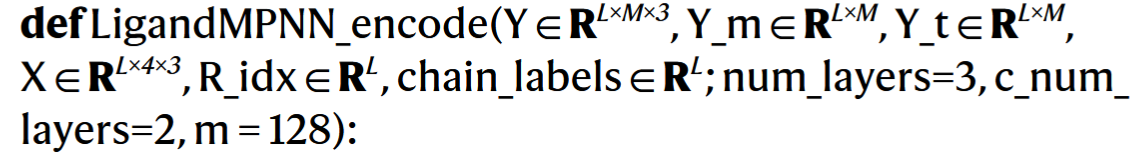
| 参数 | 含义 |
|---|---|
Y | 配体原子坐标([L×M×3]) |
Y_m | 配体掩码([L×M]) |
Y_t | 配体原子类型编号([L×M]) |
X | 蛋白质主链坐标([L×4×3]) |
R_idx | 残基编号([L]) |
chain_labels | 蛋白链 ID([L]) |
num_layers | 蛋白–蛋白图编码器层数 |
c_num_layers | 配体图编码器层数 |
m | 特征维度大小(默认 128) |
# 1. 输入数据的特征提取
 调用算法10得到,以下特征
调用算法10得到,以下特征
| 名称 | 维度 | 说明 |
|---|---|---|
v_y | [L×M×m] | 蛋白–配体图中配体节点表示 |
e | [L×K×m] | 蛋白质残基图中的边特征 |
e_idx | [L×K] | 残基邻接索引 |
Y_nodes | [L×M×m] | 配体图中原子节点特征 |
Y_edges | [L×M×M×m] | 配体图中边特征(全连接) |
# 2. 蛋白主链图编码(得到蛋白质图的边特征和节点特征)
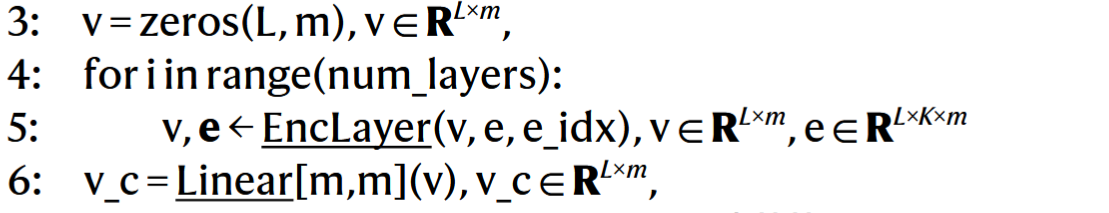
v是蛋白节点(残基)的主链初始化向量,e是边特征。- 使用多层
EncoderLayer对蛋白残基图进行消息传播。
# 3. 配体图的节点和边特征预处理
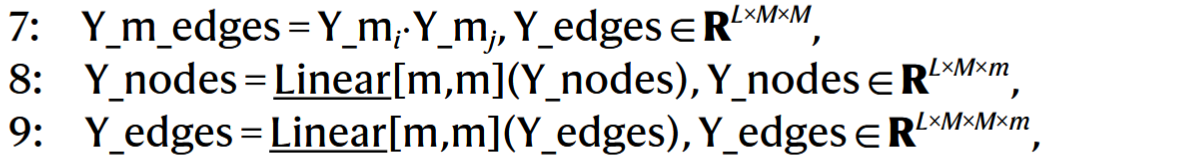
Y_m是掩码,看哪些配体是真实存在的。
# 4. 蛋白–配体图的融合

DecLayerJ是在每个残基下的配体原子子图中进行图消息传递,更新节点特征,让配体原子之间交流。v_y蛋白–配体图中配体节点表示,将配体图中的配件节点表示Y_nodes,拼接到一起得到新的配体节点表示Y_nodes_c。(感觉有点象融合蛋白-配体图和配体图)v_c蛋白图中节点(残基)的向量,将配体节点的表示Y_nodes_c看作边,使用DecLayer更新蛋白图的节点。(感觉像融合三种图)
[!note] 注
DecLayerJ(算法9,Context Decoder Layer,):这个是LigandMPNN新提出来的,主要用于在“配体原子图”中进行节点信息更新。不过和Decoder Layer很类似。DecLayer( 算法8,Decoder Layer):和proteinMPNN类似的,给出节点特征和边特征来更新节点特征。
# 5. 输出
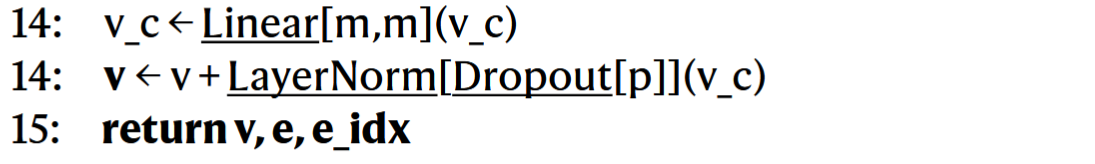
v ∈ ℝ^{L×m}:更新后的蛋白残基节点特征;(三方融合后的边特征)e ∈ ℝ^{L×K×m}:残基–残基图边特征(没有更新过);e_idx ∈ ℝ^{L×K}:图连接信息,top-K。
# 5. LigandMPNN decode function
 该函数主要实现基于蛋白–配体结构图嵌入的自回归氨基酸序列预测,输出每个残基的 21 类氨基酸的对数概率分布。
该函数主要实现基于蛋白–配体结构图嵌入的自回归氨基酸序列预测,输出每个残基的 21 类氨基酸的对数概率分布。
# 0. 函数定义
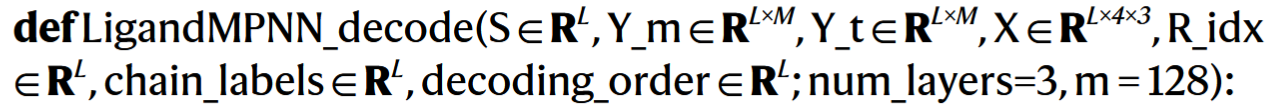
| 参数 | 类型 | 含义 |
|---|---|---|
S ∈ ℝ^L | int 序列 | 当前生成的氨基酸序列 |
Y_m ∈ ℝ^{L×M} | mask | 残基附近 M 个配体原子的掩码 |
Y_t ∈ ℝ^{L×M} | int | 配体原子类型 |
X ∈ ℝ^{L×4×3} | 坐标 | 蛋白质主链坐标(N, Cα, C, O) |
R_idx ∈ ℝ^L | int | 残基编号(用于位置编码) |
chain_labels ∈ ℝ^L | int | 链编号 |
decoding_order ∈ ℝ^L | int | 自回归生成顺序 |
num_layers | int | 解码器层数 |
m | int | 嵌入维度(如 128) |
# 1. 调用上面的算法11得到蛋白质图的边特征和节点特征
 调用 LigandMPNN_encode 融合三种图,得到融合后的蛋白质图的边特征和节点特征。
调用 LigandMPNN_encode 融合三种图,得到融合后的蛋白质图的边特征和节点特征。
# 2. 构建自回归mask,
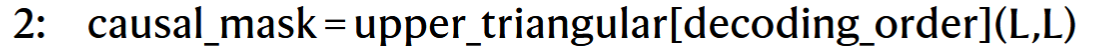
- 按照
decoding_order创建上三角掩码; - 只允许“已生成的位置”影响“当前待生成位置”。
# 3. 将当前序列 S 嵌入为节点特征和边特征
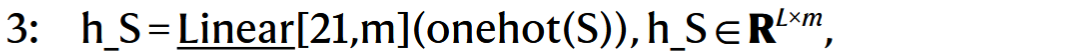
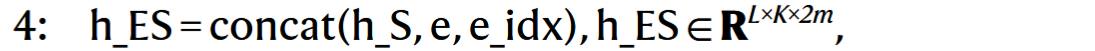
h_S表示已预测的序列S的残基节点特征- 通过拼接节点特征和蛋白图的边特征和位置编码得到当前序列的边特征
h_ES。
# 4. 构造替代边特征(用于 masked 位置,没见过预测序列的边表示)
 与
与h_ES区别是,前面使用零填充,表示不会看见当前已预测的序列特征。
# 5. 融合蛋白质图的节点表示和已预测序列无关的边表示
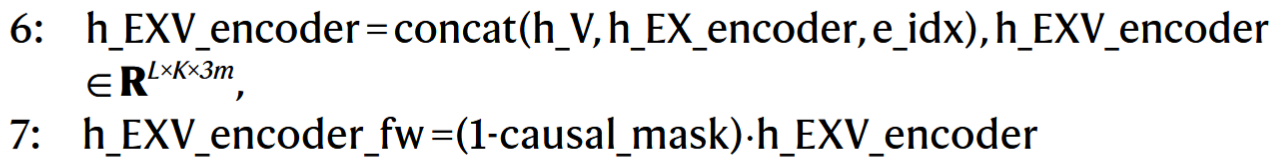 融合就是拼接。然后使用
融合就是拼接。然后使用causal_mask来控制哪些位置可以“看见”其他残基的氨基酸类型。
# 6. 使用DecLayer迭代更新带有当前预测序列的边特征

- 融合蛋白质图的节点表示和已预测序列有关的边表示,得到新的边表示
- 根据
causal_mask,融合无关和有关的边表示 - 根据已预测的序列表示更新蛋白质图的节点表示。
# 7. 输出预测的氨基酸概率
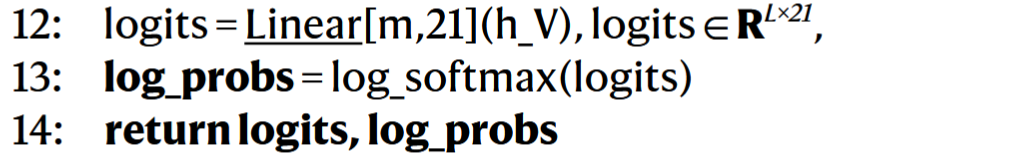
# 6. Outline of LigandMPNN sidechain decode function 侧链构象生成模块
 对每个残基,预测 四个 χ 角(chi1~chi4) 的概率分布,每个角度建模为三分量 Von Mises 分布混合模型(Mixture of Von Mises)。并没有使用简单的回归预测角度值
对每个残基,预测 四个 χ 角(chi1~chi4) 的概率分布,每个角度建模为三分量 Von Mises 分布混合模型(Mixture of Von Mises)。并没有使用简单的回归预测角度值
[!note] 背景知识:侧链 χ 角 & Von Mises 分布
- 每个氨基酸残基的侧链由 1 到 4 个扭转角(χ₁~χ₄)决定;
- 它们是 圆形变量,值域是 ;
- 用正态分布建模会忽略角度的周期性,而 Von Mises 分布(环状高斯)非常适合建模这种类型;