 DeepGO-SE
DeepGO-SE
# Protein function prediction as approximate semantic entailment
Journal: Nature Machine Intelligence (IF 23.9) Published: 2024年2月14日 开源地址:https://github.com/bio-ontology-research-group/deepgo2
# GO: Gene Ontology
基因本体论(Gene Ontology,简称GO)是一个大型的国际合作生物信息学项目。它的核心目标是创建一个标准化的、动态更新的、可被计算机理解的词汇体系,用来全面地描述基因和蛋白质在各个生物体中所扮演的角色。 简单来说,GO提供了一套“标准语言”,让全世界的科学家能够用统一、精确的方式来描述基因和蛋白质的功能,从而方便数据的交换、比较和分析。
- biological_process (GO:0008150) [生物学过程]
- cellular process (GO:0009987) [细胞过程]
- cell death (GO:0008219) [细胞死亡]
- programmed cell death (GO:0012501) [程序性细胞死亡]
- is_a -> apoptotic process (GO:0006915) [细胞凋亡过程]
2
3
4
5
这个结构清晰地显示了“细胞凋亡过程”是一个非常具体的概念,它从属于“程序性细胞死亡”,而“程序性细胞死亡”又从属于更宽泛的“细胞死亡”等概念。
# Gene Ontology的核心组成部分
GO通过三个独立的、但又相互关联的本体(Ontology)或领域(Domain)来描述基因产物(即蛋白质或RNA)的生物学知识。 这三个领域分别是:
分子功能 (Molecular Function, MFO):
- 定义:描述基因产物在分子层面上的“工作”或“活动”。 这指的是基因产物本身具备的能力,例如与什么分子结合,或者催化什么化学反应。
- 特点:这个领域的描述不涉及具体的时间、地点或与其他分子的协作。它只关注该分子“能做什么”。
- 例子:“催化活性” (catalytic activity)、“DNA结合” (DNA binding) 或“转运蛋白活性” (transporter activity)。
生物学过程 (Biological Process, BPO):
- 定义:描述由一个或多个分子功能有序组合而成的、更大范围的生物学事件或目标。 它回答的是基因产物参与了“哪个生物学故事”。
- 特点:一个生物学过程通常需要多个基因产物的协同作用才能完成。
- 例子:“DNA修复” (DNA repair)、“细胞信号传导” (signal transduction) 或“新陈代谢” (metabolic process)。一个参与“DNA修复”的蛋白质,其“分子功能”可能是“DNA结合”和“核酸内切酶活性”。
细胞组分 (Cellular Component, CCO):
- 定义:描述基因产物在细胞中所处的位置,或者说它是哪个细胞结构的组成部分。 它回答的是基因产物“在哪里工作”。
- 特点:这包括了从大的细胞器到微小的蛋白质复合物。
- 例子:“细胞核” (nucleus)、“线粒体” (mitochondrion)、“细胞质” (cytoplasm) 或“核糖体” (ribosome)。
# Gene Ontology的结构特点
GO不仅仅是一个简单的词汇列表,它具有严谨的逻辑结构:
- 层级结构:GO的词条(GO term)之间存在明确的父子关系,形成一个有向无环图(Directed Acyclic Graph, DAG)的结构。 这意味着一个具体的词条(子词条)可以从属于一个或多个更宽泛的词条(父词条)。例如,“线粒体内膜”是“线粒体膜”的子集,也是“内膜系统”的子集。
- “真路径规则” (True Path Rule):如果一个基因产物被注释到了某个GO词条,那么它也必然属于该词条的所有上级(父)词条。这使得研究人员可以根据需要,在不同详细程度上进行功能分析。
# Gene Ontology的应用
GO在现代生物学和生物信息学研究中扮演着至关重要的角色,其主要应用包括:
- 功能注释 (Functional Annotation):科学家们通过实验数据,将基因和蛋白质与最能描述其功能的GO词条关联起来。这个过程称为“功能注释”。
- 功能富集分析 (Functional Enrichment Analysis):这是GO最常见的应用之一。例如,当研究人员通过高通量实验(如基因芯片或转录组测序)得到一个包含上百个差异表达基因的列表时,他们可以使用GO来分析这些基因主要集中在哪些生物学过程或分子功能上,从而揭示实验处理可能导致的生物学变化。
- 模型生物数据整合:由于GO的标准化特性,它可以用来比较不同物种(如人类、小鼠、酵母)的基因功能,促进了知识的迁移和整合。
- 构建知识库:GO是构建大型生物学知识库(如UniProtKB/Swiss-Prot)的基石,为自动化功能预测和系统生物学研究提供了支持。
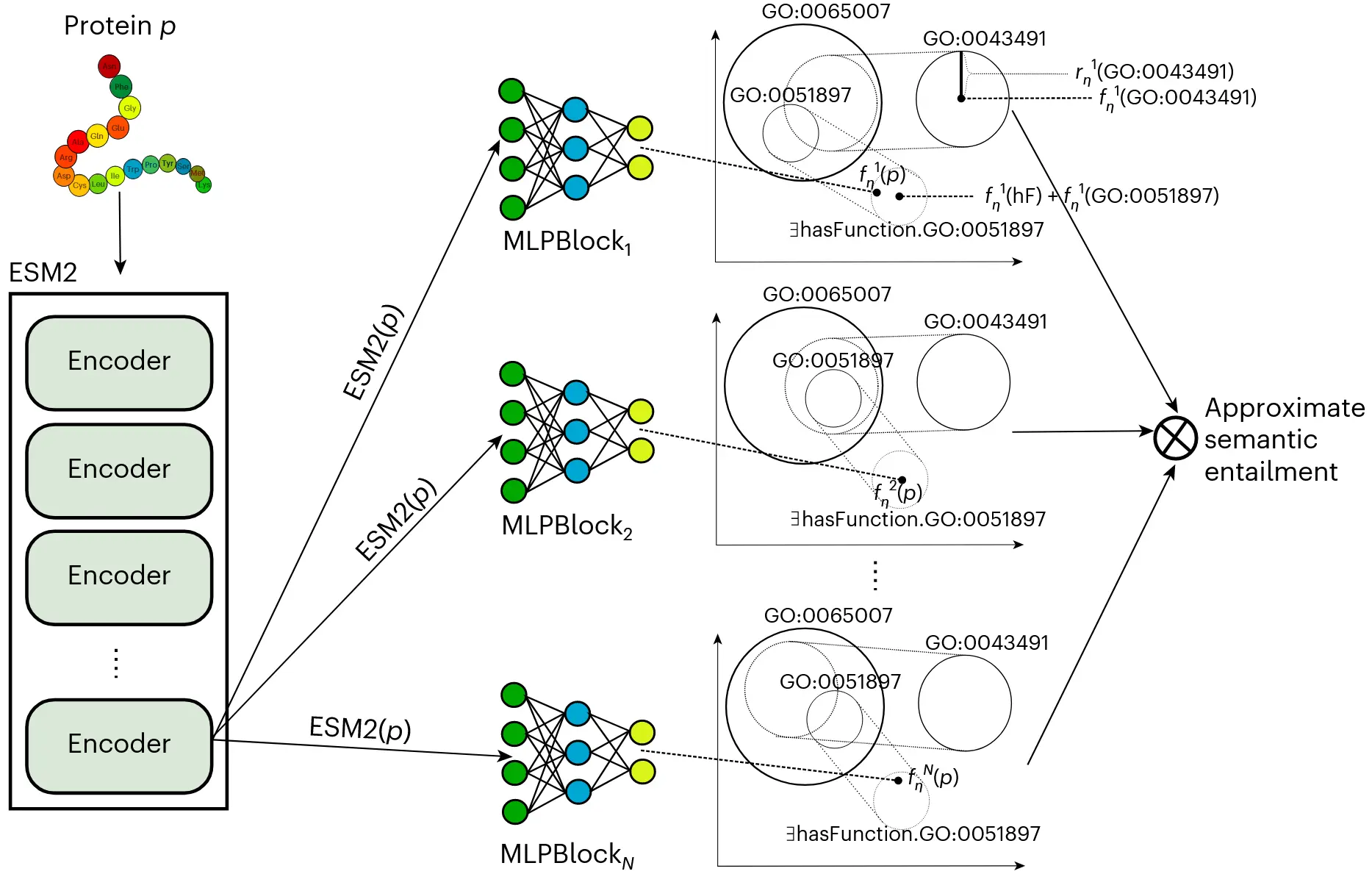
# 模型结构
# 结构:
DeepGO-SE是一个端到端的神经-符号混合模型。其核心机制可以分解为以下几个部分:
# 1. 蛋白质序列的向量化表示
模型采用 ESM2 (Evolutionary Scale Modeling 2),一个基于Transformer架构的大型语言模型。
- 输入:蛋白质的氨基酸序列。
- 输出:一个能够捕捉蛋白质深层生物化学、结构和进化信息的特征向量
ESM2(p)。
# 2. GO知识库的几何化表示:ELEmbeddings
为了让神经网络能够“理解”GO的逻辑公理,论文采用了ELEmbeddings技术。
- 原理:该技术将描述逻辑本体(如GO)嵌入到一个欧几里得空间中。
- 实现:
- 每个GO术语(class)
C被表示为一个n维球体(n-ball),由其中心向量c和半径r(c)定义。 - GO的公理被翻译成几何约束。例如,公理
C ⊑ D(C是D的子类) 被建模为要求C的n维球体必须完全包含在D的n维球体之内。 - 这些几何约束被转化为一个可微的损失函数(loss function),可以在模型训练过程中进行优化,从而迫使模型学习到一个在几何上满足GO逻辑的表示空间。
- 每个GO术语(class)
# 3. 核心机制:近似语义蕴含
这是论文最具创新性的部分,直接对应其标题。
- 语义蕴含 (Semantic Entailment) 的定义:在逻辑学中,一个理论
T(此处指GO的所有公理)语义上蕴含一个陈述φ(此处指“蛋白质p具有功能c”),记为T ⊨ φ,当且仅当在所有使T为真的模型(即解释,interpretation)中,φ也为真。 - 计算上的挑战:一个理论的模型集合
Mod(T)通常是无限的,因此直接计算语义蕴含是不可行的。 - DeepGO-SE的近似方案:
- 学习有限模型集合:模型不尝试遍历无限的模型空间,而是通过
N个并行的多层感知机(MLP)模块(MLPBlock)学习生成N个不同的、但都近似满足GO公理的有限模型/世界{ℐ₁, ℐ₂, ..., ℐₙ}。 - 在每个模型中进行预测:对于一个蛋白质
p,其ESM2嵌入被分别输入到这N个MLP中,投影到N个不同的几何表示空间。在每个空间ℐᵢ中,模型计算陈述hasFunction(p, c)的真值(truth value)。在几何上,这对应于蛋白质p的投影向量fᵢ(p)落入GO术语c的n维球体Bᵢ(c)的概率。 - 聚合真值:最终的预测分数是对这
N个模型各自预测的真值进行聚合的结果。论文探索了不同的聚合函数,如min、max和mean。当聚合函数为min时,意味着只有当一个预测在所有N个学习到的模型中都具有高置信度时,最终的预测分数才会高。这个过程在计算上模拟了“在所有模型中为真”这一语义蕴含的核心思想。
- 学习有限模型集合:模型不尝试遍历无限的模型空间,而是通过
# 训练
# 准备工作与输入数据
在开始训练之前,需要准备好以下四类数据:
- 蛋白质序列数据:一个包含所有训练蛋白质氨基酸序列的文件。
- 真实功能注释(Ground Truth Labels):蛋白质与其对应的GO术语的关联。对于一个包含
M个GO术语的子本体(如MFO),每个蛋白质的标签是一个M维的向量(multi-hot vector)Y_true。如果蛋白质p具有第j个GO功能,则Y_true[j] = 1,否则为0。 - GO本体文件(Ontology File):这是包含GO知识库结构的文件,通常是OWL或OBO格式。关键是,它不仅定义了GO术语,还包含了它们之间的逻辑公理(axioms),例如
GO:A ⊑ GO:B(A是B的子类)。
# 训练循环(Training Loop)中的核心步骤
对于训练集中的每一个数据批次(batch),模型都会执行以下四个步骤:
# 步骤一:前向传播 (Forward Pass) — 生成预测
序列编码 (Sequence Encoding):
- 从批次中取出一个蛋白质
p的氨基axit序列。 - 将该序列输入到预训练好且已冻结(frozen)的ESM2模型中。这意味着在训练DeepGO-SE时,ESM2模型的参数是固定不变的,它仅作为特征提取器。
- ESM2输出一个高维的特征向量
esm2(p),该向量是对蛋白质序列的浓缩表示。
- 从批次中取出一个蛋白质
多模型投影 (Multi-model Projection):
- 模型内部有
N个并行的、结构相同但参数独立的多层感知机(MLPBlock)。 - 将上一步得到的向量
esm2(p)同时输入到这N个MLPBlock中。 - 每个
MLPBlockᵢ将esm2(p)投影到它自己负责的那个几何表示空间中,生成N个不同的蛋白质表示向量:{f₁(p), f₂(p), ..., fₙ(p)}。每一个fᵢ(p)都代表蛋白质p在第i个“可能世界”或“逻辑模型”中的坐标。
- 模型内部有
多模型评分 (Multi-model Scoring):
- 对于子本体中的每一个GO术语
c,模型需要在每一个(共N个)几何空间中计算其与蛋白质p的关联分数。 - 在第
i个模型中,分数Sᵢ(p, c)由以下公式计算:Sᵢ(p, c) = σ( dot(fᵢ(p), fᵢ(hF) + fᵢ(c)) + rᵢ(c) )fᵢ(p)是蛋白质p在模型i中的向量。fᵢ(c)和rᵢ(c)是GO术语c在模型i中对应的n维球体的可学习的中心向量和半径。fᵢ(hF)是“hasFunction”这个关系在模型i中的可学习的嵌入向量。dot()是点积运算,σ是Sigmoid激活函数,将分数归一化到 (0, 1) 区间,使其具有概率意义。
- 对于子本体中的每一个GO术语
分数聚合 (Score Aggregation) — 近似语义蕴含:
- 经过上一步,对于每一个蛋白质-功能对
(p, c),我们得到了N个分数{S₁(p, c), S₂(p, c), ..., Sₙ(p, c)}。 - 模型使用一个预定义的聚合函数(如
min,max, 或mean)将这N个分数聚合成一个最终的预测分数Ŷ(p, c)。例如,如果使用min聚合,则Ŷ(p, c) = min(S₁(p, c), ..., Sₙ(p, c))。 - 对所有GO术语
c执行此操作后,得到最终的预测向量Ŷ,其维度与真实标签Y_true相同。
- 经过上一步,对于每一个蛋白质-功能对
# 步骤二:损失计算 (Loss Calculation) — 衡量误差
模型的总损失由两个独立的部分加权构成:
A. 预测损失 (Prediction Loss, L_pred)
- 目的:衡量模型的预测与真实功能注释之间的差距。
- 计算:使用二元交叉熵损失函数(Binary Cross-Entropy Loss, BCE Loss)。这是处理多标签分类问题的标准损失函数。
L_pred = BCE(Ŷ, Y_true) - 这个损失会驱动模型学习生成能够匹配真实数据的预测。
B. 几何损失 (Geometric Loss, L_geom)
- 目的:强制模型内部的
N个几何表示空间遵守GO知识库中的逻辑公理。 - 计算:
- 遍历GO本体文件中的每一条公理(例如,
'GO:A' ⊑ 'GO:B')。 - 对于每一条公理,在每一个(共
N个)几何模型中,计算其“违背程度”(violation)。例如,对于公理'A' ⊑ 'B'在模型i中的违背程度是max(0, ||cᵢ(A) - cᵢ(B)|| + rᵢ(A) - rᵢ(B))。 L_geom是所有公理在所有N个模型中的违背程度之和。
- 遍历GO本体文件中的每一条公理(例如,
- 这个损失是一个正则化项,它不关心预测是否准确,只关心模型内部的知识表示是否符合逻辑。
C. 总损失 (Total Loss, L_total)
- 将上述两个损失加权求和:
L_total = L_pred + α * L_geom α是一个超参数,用于平衡预测准确性和逻辑一致性的重要性。
# ELEmbeddings
看看 ELEmbeddings 是如何实现“GO知识库的几何化表示”的。为了理解ELEmbeddings,我们首先要明确它试图解决的根本问题:
根本问题:神经网络(Neural Networks)的世界是连续的、数字化的(由向量和矩阵构成),而基因本体论(Gene Ontology, GO)这样的知识库是符号化的、离散的(由概念和逻辑规则构成)。这两者之间存在一道鸿沟。我们如何让一个只会做数学计算的神经网络,“理解”并“遵守”像“如果A是B的子类,那么拥有功能A的蛋白质必然拥有功能B”这样的逻辑规则呢?
ELEmbeddings 将逻辑规则翻译成几何关系。
# ELEmbeddings 技术讲解
ELEmbeddings 的核心思想可以分解为两个部分:
- 如何表示一个“概念” (GO术语/Class)?
- 如何表示它们之间的“逻辑规则” (GO公理/Axiom)?
# 第一部分:用几何形状表示“概念”
在传统的词嵌入(如Word2Vec)中,一个词被表示为空间中的一个点(一个向量)。但一个点无法表达一个概念的“范围”或“不确定性”。
ELEmbeddings 做了改进:它将每一个GO术语(如“细胞核”、“催化活性”)表示为高维空间中的一个 n维球体 (n-ball)。
一个n维球体由两个参数定义:
- 中心点向量 (Center vector)
c:这个向量捕捉了该概念的核心语义。可以把它想象成这个概念最典型的“意思”。 - 半径 (Radius)
r:这是一个标量值,代表了这个概念的“范围”或“覆盖面”。一个非常宽泛的概念(如“细胞器”)可能会有一个较大的半径,而一个非常具体的概念(如“线粒体内膜”)则会有一个较小的半径。
所以,在ELEmbeddings中:
- “细胞核” 不再是一个点,而是空间中的一个球体
Ball(c_核, r_核)。 - “细胞器” 也是一个球体
Ball(c_器, r_器)。
# 第二部分:用几何约束表示“逻辑规则”
这是ELEmbeddings最关键的一步。它将GO知识库中的逻辑公理,翻译成了对这些n维球体的位置和大小的几何约束。
我们以GO中最常见、也最重要的一条逻辑规则为例:子类关系 (Subclass of),记为 C ⊑ D (C是D的子类)。
逻辑陈述:
'线粒体' ⊑ '细胞器'(线粒体是一种细胞器)。几何翻译:如果“线粒体”是“细胞器”的子类,那么代表“线粒体”的球体,必须完全被代表“细胞器”的球体所包含。
如何用数学来表达这个几何约束? 一个球体A被另一个球体B包含,需要满足一个条件:A的中心点和B的中心点之间的距离,加上A的半径,必须小于或等于B的半径。 用公式表达就是:
|| c_A - c_B || + r_A ≤ r_B其中||...||代表向量的欧几里得距离(即两个中心点之间的直线距离)。
GO知识库中包含成千上万条这样的公理。ELEmbeddings会把每一条公理都转换成类似上述的几何约束方程。例如:
C ⊑ D->||c_C - c_D|| + r_C ≤ r_DC ⊓ D ⊑ E(C和D的交集是E的子类) -> 也有对应的、更复杂的几何约束。
# 如何让神经网络“遵守”这些规则?——通过损失函数
现在我们有了数学化的几何约束,但神经网络在训练开始时,所有这些球体的位置和半径都是随机初始化的,完全不满足这些约束。如何迫使它们在训练过程中逐渐“排列整齐”,最终满足GO的逻辑结构呢?
答案是:设计一个“惩罚”项,即几何损失函数 (Geometric Loss Function)。
定义“违规度” (Violation):对于每一条公理,比如
'线粒体' ⊑ '细胞器',模型会计算当前的几何表示“违背”这条规则的程度。 根据上面的公式||c_线粒体 - c_细胞器|| + r_线粒体 ≤ r_细胞器,我们可以定义一个“违规度”函数:Loss_axiom = max(0, ||c_线粒体 - c_细胞器|| + r_线粒体 - r_细胞器)理解这个损失函数:
- 如果约束被满足:
||...|| + r_线粒体 - r_细胞器的结果将是一个负数或0。那么max(0, ...)的结果就是0。这意味着模型没有违反规则,不产生任何惩罚 (Loss=0)。 - 如果约束被违反:
||...|| + r_线粒体 - r_细胞器的结果将是一个正数。这个正数的大小,就精确地量化了“线粒体”球体“戳出”细胞器球体的程度。这个正数就是惩罚值 (Loss > 0)。
- 如果约束被满足:
整合到总训练目标中: 模型在训练时,其总的损失函数 (Total Loss) 由两部分组成:
Total Loss = Prediction Loss + α * Geometric Loss- Prediction Loss:这是标准的监督学习损失,用于衡量模型的预测与真实标签之间的差距(例如使用交叉熵损失)。它驱使模型做出准确的预测。
- Geometric Loss:这是所有GO公理产生的“违规度”惩罚的总和。它驱使模型调整所有n维球体的中心点
c和半径r,使得它们之间的几何关系尽可能地满足整个GO知识库的逻辑结构。 α是一个超参数,用于平衡这两个目标的重要性。
训练过程就像一个双重任务:模型一方面要努力学习如何正确预测蛋白质功能(最小化Prediction Loss),另一方面,它必须在“整理”内部的几何知识库,让所有概念球体各就其位,遵守逻辑规则(最小化Geometric Loss)。
经过训练后,ELEmbeddings为我们创造了一个结构化的、语义丰富的向量空间。在这个空间里:
- 向量(球体的中心)的位置不再是任意的,它们的相对位置和包含关系反映了GO的内在逻辑。
- 当DeepGO-SE模型将一个蛋白质的ESM2向量投影到这个空间时,它实际上是在这个已经“懂”了生物学逻辑的坐标系中为该蛋白质寻找一个定位。如果蛋白质的向量落在了“线粒体”球体内,那么根据“真路径规则”(True Path Rule),它也必然位于“细胞器”球体内。这样,模型的预测就天然地具有了逻辑一致性。