 GraphEC
GraphEC
# Accurately predicting enzyme functions through geometric graph learning on ESMFold-predicted structures
Journal:Nature CommunicationsIF 15.7 Published: 2024年9月18日 开源地址:https://github.com/biomed-AI/GraphEC
# 模型架构
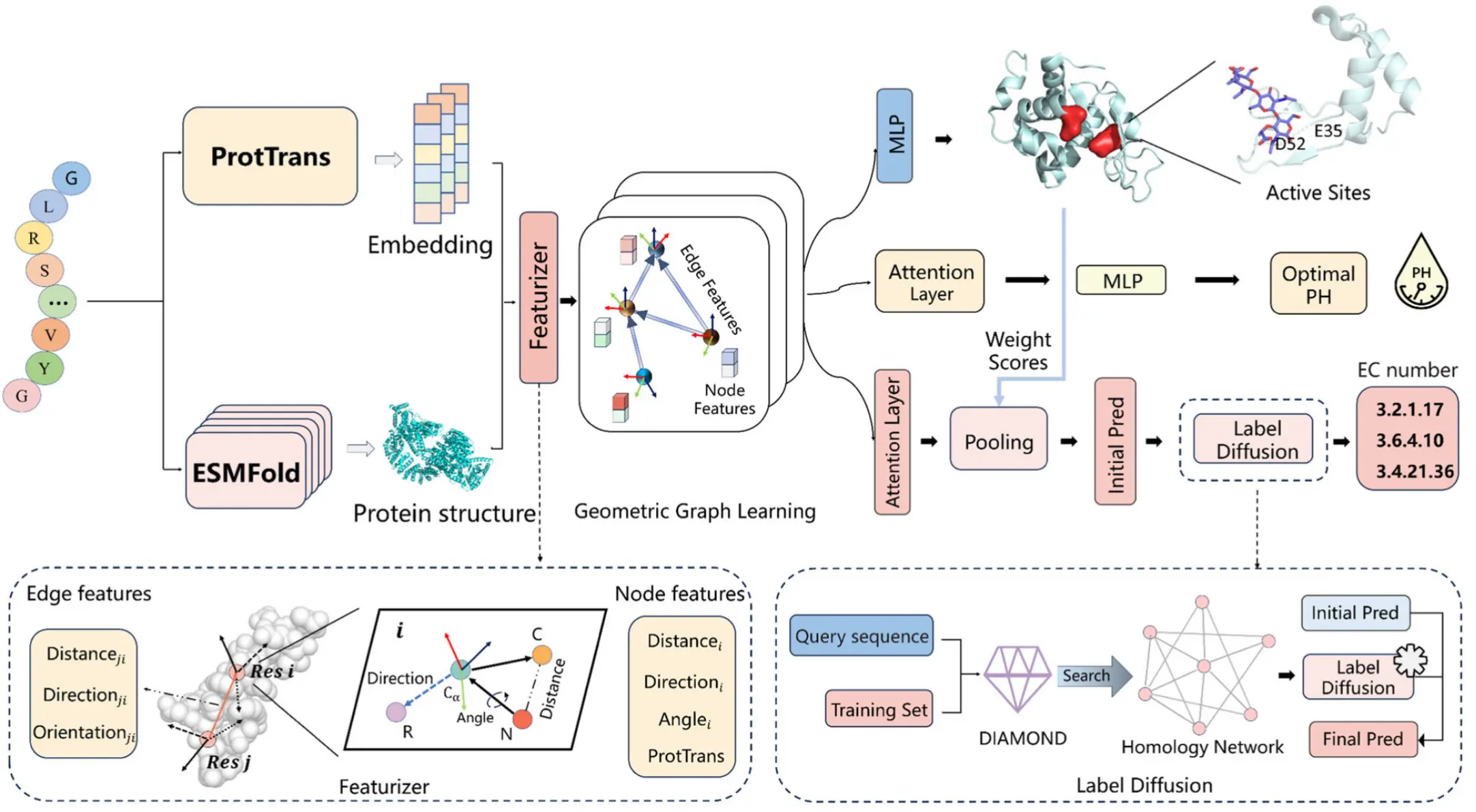
GraphEC是一个基于几何图学习的端到端框架,旨在从蛋白质的一级序列预测其EC编号、活性位点及最适pH值。其核心架构整合了三个主要部分:1) 基于ESMFold的快速结构预测和图构建;2) 一个用于学习结构-功能关系的几何图神经网络(GNN);3) 针对不同预测任务的特定输出模块(Task-Specific Heads),并通过标签扩散算法进行后处理优化。
# 第一步:结构预测与图表示 (Structure Prediction & Graph Representation)
输入: 蛋白质的一级氨基酸序列 (Protein Sequence)。
结构预测: 利用蛋白质语言模型 ESMFold 对输入序列进行三维结构预测。ESMFold能够以比AlphaFold2快数十倍的速度生成具有原子级分辨率的结构坐标,同时保持了较高的准确性,这使其适用于高通量分析。
图构建 (Graph Construction): 将预测出的三维结构转换为图 。
- 节点 (Vertices, V): 每个氨基酸残基被定义为一个节点 。通常使用其α-碳原子 (Cα) 的坐标 作为节点的空间位置。
- 边 (Edges, E): 采用 半径图 (Radius Graph) 的构建方式。如果两个节点 和 的空间欧氏距离小于预设的半径阈值 (例如, 10Å),即 ,则在它们之间创建一条边 。
# 第二步:节点与边的特征工程 (Node & Edge Featurization)
为图中的节点和边赋予丰富的初始特征,这些特征是后续学习的基础。
节点特征 (h_i): 每个节点 的初始特征向量 由两部分拼接而成:
- 几何特征: 为了捕捉局部三维环境,首先为每个残基 构建一个局部坐标系 ,该坐标系由其主链原子N, Cα, C的位置确定。基于此坐标系,计算残基内部原子间(如Cα-C, Cα-N等)以及与侧链质心之间的距离、方向和角度。距离特征通过径向基函数 (Radial Basis Functions, RBF)进行编码。
- 序列特征: 使用预训练的蛋白质语言模型 ProtTrans (ProtT5-XL-U50),为每个残基生成一个高维嵌入向量。该嵌入向量捕获了在海量蛋白质序列数据中学习到的进化信息和上下文语境。
边特征 (e_ij): 每条边 的特征向量 描述了两个相连残基 和 之间的空间关系:
- 距离与方向: 类似于节点特征,计算残基 和 之间不同原子对的距离(经RBF编码)和方向向量。
- 相对方向 (Orientation): 这是一个关键的几何特征。它通过计算两个局部坐标系 和 之间的相对旋转来获得,通常表示为一个四元数 q()。这使得模型能够理解一个残基相对于另一个残基的精确空间朝向。
# 第三步:几何图学习 (Geometric Graph Learning)
此步骤是模型的核心,通过一个多层的几何图神经网络(GNN)来学习和更新节点表示。
信息传播机制: 该GNN采用基于多头注意力机制 (Multi-Head Attention) 的消息传递框架。在第 层,节点 的特征向量 会通过聚合其邻居节点 的信息进行更新。
节点更新 (Node Update): 节点 的特征从 更新到 的过程可以形式化地表示为:
其中, 是从节点 发送到节点 的“消息”,其计算方式如下:
- 和 是可学习的权重矩阵,分别用于变换节点和边的特征。
- 是注意力权重,通过节点 的查询向量 (Query) 和节点 的键向量 (Key) 的点积计算得到,表示节点 对邻居 的关注程度。这使得模型能动态地学习哪些邻居信息更重要。
边更新 (Edge Update): 在节点更新后,边的特征也相应更新,以反映节点状态的变化:
其中 表示拼接操作,EdgeMLP是一个多层感知机。
经过多层GNN的迭代,每个节点最终得到一个几何嵌入向量 (Geometric Embedding),该向量高度浓缩了其局部化学和物理环境以及在整个蛋白质结构中的功能性上下文信息。
# 第四步:任务特定的预测模块 (Task-Specific Prediction Heads)
从GNN获得的最终几何嵌入被送入不同的下游模块以完成具体预测任务。
酶活性位点预测 (GraphEC-AS):
- 架构: 将每个残基的最终几何嵌入向量输入到一个独立的多层感知机 (MLP) 中。
- 输出: 对每个残基输出一个标量值,该值通过Sigmoid函数转换为0到1之间的概率,表示该残基是活性位点的可能性。
- 作用: 此模块不仅是一个独立的预测任务,其输出的概率分数还将作为注意力引导,用于指导后续的EC编号预测。
EC编号预测 (GraphEC):
- 注意力加权池化 (Attention-Weighted Pooling): 为了从残基级别的嵌入得到蛋白质级别的表示,模型采用了一种加权池化策略。每个残基的几何嵌入向量会根据其在GraphEC-AS模块中得到的“活性位点分数”进行加权。这使得模型在聚合信息时,能重点关注功能上更关键的区域。
- 初始预测: 聚合后的蛋白质级别向量通过一个MLP分类器,得到一个关于所有EC编号的初始概率分布向量 。这是一个多标签分类问题。
- 后处理:标签扩散算法 (Label Diffusion Algorithm): 在推理阶段,为提高预测的鲁棒性,采用标签扩散算法对 进行优化。
- 利用DIAMOND工具为待测序列在训练集中寻找同源序列,构建一个同源网络。
- 最终的预测分数 通过求解一个优化问题得到,其目标是平衡初始预测的保真度和同源网络中标签的一致性。该解具有闭式形式:
其中, 是包含初始预测的矩阵, 是单位矩阵, 是同源网络的图拉普拉斯矩阵, 是正则化参数。该过程有效地将高置信度的同源蛋白标签信息“扩散”到待测序列上,从而校正和增强预测结果。
最适pH值预测 (GraphEC-pH):
- 架构: 类似于EC编号预测,它也通过注意力池化将残基嵌入聚合为蛋白质级向量。
- 输出: 蛋白质级向量被送入一个MLP分类器,用于预测最适pH值所属的类别(如:酸性、中性、碱性)。这是一个多类别分类任务。
通过以上步骤,GraphEC模型实现了从单一序列输入到多维度、高精度酶功能注释的完整流程。