 MODIFY
MODIFY
# MODIFY: Machine learning-guided co-optimization of fitness and diversity facilitates combinatorial library design in enzyme engineering
nature communications 2024
# 背景
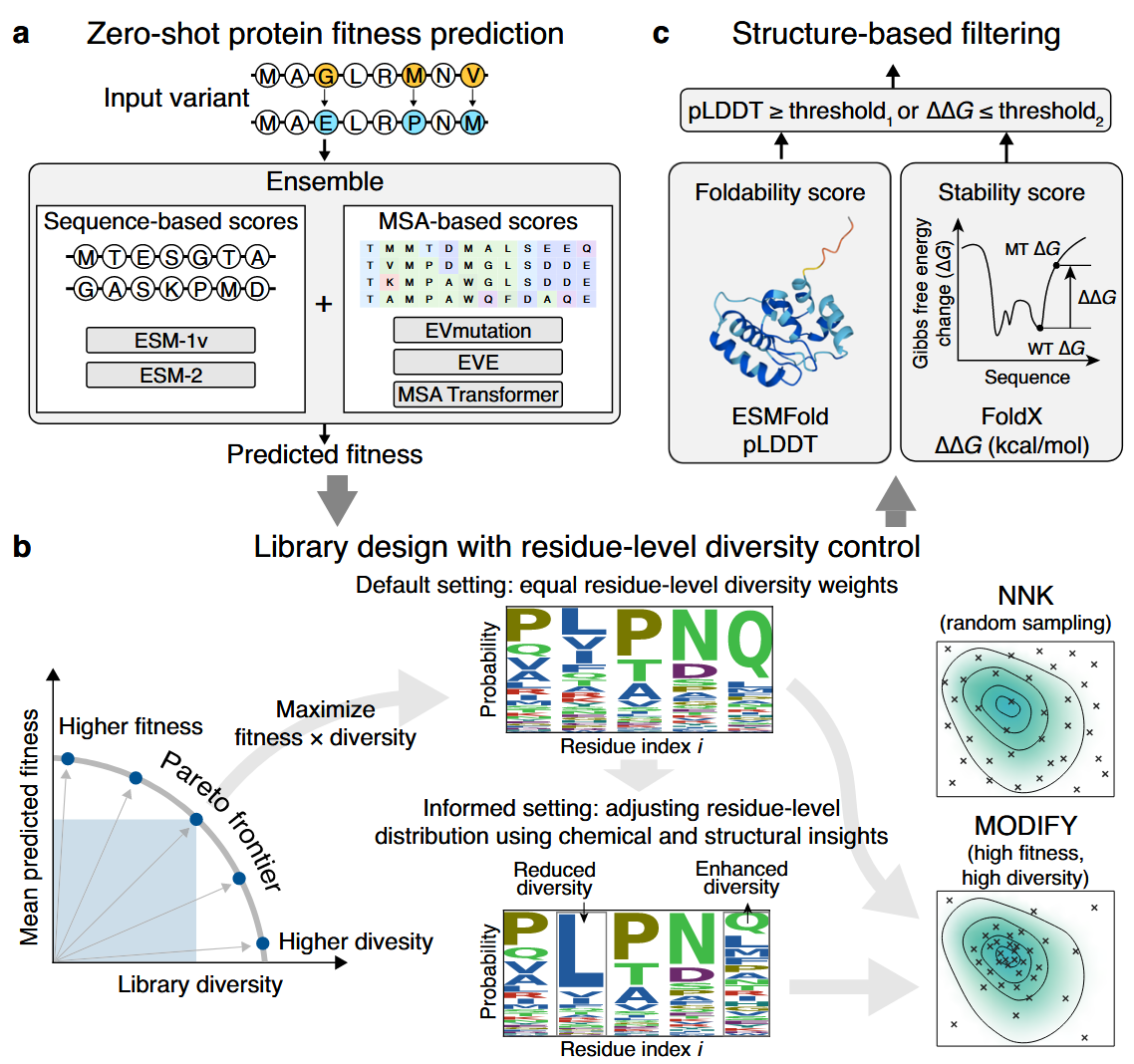
在酶工程中,设计一个高质量的起始突变库对发现功能性变体至关重要。MODIFY解决的问题就是如何在没有已知实验数据的情况下,设计出既有功能性(适应度、合理性、fitness) 又 具多样性(diversity) 的突变组合。 MODIFY目标就是同时最大化酶变体的预测功能性与序列多样性。
- 零样本预测(Zero-shot fitness prediction):利用多个无监督模型(ESM-1v、ESM-2、EVmutation、EVE、MSA Transformer)组成的集成模型预测蛋白质变体的功能性。
- 帕累托最优( Pareto Optimality):设计使功能性与多样性之间达到最优权衡的突变库。
# 零样本预测(Zero-shot fitness prediction)
MODIFY 利用 4 种模型来对 fitness 进行评分
# 1. 蛋白语言模型(Protein Language Model, PLM)
为实现零样本适应性预测,MODIFY 利用了预训练的 蛋白语言模型(ESM),通过对天然蛋白序列中氨基酸的上下文关系建模,推断突变是否合理,进而作为变体适应性的代理指标。
# 建模目标
给定一个蛋白序列 ,其中:
- 是 20 种标准氨基酸构成的字母表;
- 是序列长度。 蛋白语言模型的核心任务是估计序列中某一位点氨基酸的条件概率:
其中 表示除了位置 以外的序列上下文。 为了评估一个突变体序列 的可行性,MODIFU计算其与野生型序列 在突变位点上的 对数几率比(log-odds ratio):
- :所有发生突变的位点;
- :将突变位点掩蔽后的序列上下文;
- 当 时,表示突变体在进化上更可行,从而可能具有更高的适应性。
# 2. 进化耦合模型(Evolutionary Coupling Model)
除了 ESM 捕捉到的全局进化上下文信息外,还引入了进化耦合模型,用于捕捉特定蛋白的局部进化上下文,即来源于该蛋白的同源序列。通过考虑蛋白序列中的单个位点偏好与共进化模式(残基间协同变异) 来学习一个序列 的“能量”函数 :
- :残基位置 上氨基酸 的偏好;
- :残基对 上氨基酸组合的共进化耦合强度; 其中参数 和 是通过该蛋白的同源序列的多序列比对(MSA)进行拟合得到的。 序列的概率则定义为:
其中 是归一化常数。 使用了名为 EVmutation 的 EC 模型,并通过下面的公式来量化突变体的进化可行性:
[!note] EVmutation (Nature Biotechnology 2017) 是一种基于 MSA 的无监督蛋白突变预测模型,利用统计物理中的 Potts 模型计算序列能量,以此推测突变体是否被进化接受。
# 3. 潜变量生成序列模型(Latent Generative Sequence Model)
使用了另一类模型来引入局部的进化上下文信息,即 潜变量生成序列模型,该模型学习一个蛋白家族的序列分布。这类模型通过引入变分自编码器(VAE)模型,表达更高阶的残基依赖关系。 在本研究中,使用了 EVE (Nature 2021) 作为潜变量序列模型:
# 4. 基于多序列比对的蛋白语言模型(MSA-based PLM)
进一步引入了 MSA Transformer,虽然它与 ESM 类似,但 MSA Transformer 还额外结合了输入蛋白的同源序列多序列比对(MSA)信息,从而预测给定序列上下文中每个位点氨基酸的概率:
MSA Transformer 的评分方式 与 ESM 的评分 类似,也是通过对所有突变位点上的对数几率比(log-odds ratio) 进行求和来定义的:
# 帕累托最优( Pareto Optimality)
通过联合优化 预期功能性(fitness) 与 序列多样性(diversity),构建一个在这两者之间实现 Pareto 最优的组合库:
- :组合库的分布
- :MODIFY的预测得分
- :调节“开发性探索”(多样性)与“功能性利用”的权重
# Diversity 的定义:
使用位置级别的 熵(entropy) 表示每个位点的氨基酸多样性:
- :被设计突变的残基位点数量
- :第 个位点的多样性权重(可以调控)
- :该位点所有20种氨基酸的概率分布
- :标准氨基酸数目
- :第 位点的氨基酸分布熵
- 如果所有氨基酸概率平均(即 ),熵最大,表示多样性高;
- 如果只有一个氨基酸概率为1,其他为0,熵为0,表示多样性低。
# 帕累托边界的构建方式(关键点):

- 变动 λ 的值:从小到大(如:0 → 2)
- 小 λ:更偏向于功能性
- 大 λ:更重视多样性
- 每一个 λ 值对应一个解(一个突变库)
- 收集所有解,形成 Pareto 边界曲线
- 可以从中选择适当的权衡点
·
← LigandMPNN EpHod→